
https://en.wikipedia.org/w/index.php?curid=3925138,
accessed 9 April 2017).So far in this course we have dealt entirely either with the evolution of characters that are controlled by simple Mendelian inheritance at a single locus or with the evolution of molecular sequences. Even on Tuesday when we were dealing with population genomic data, data from hundreds or thousands of loci, we were treating the variation at each locus separately and combining results across loci. I have some old notes on gametic disequilibrium and how allele frequencies change at two loci simultaneously, but they’re in the “Old notes, no longer updated” section of the book version of these notes (https://figshare.com/articles/journal_contribution/Lecture_notes_in_population_genetics/100687), and we didn’t discuss them.1 In every example we’ve considered so far we’ve imagined that we could understand something about evolution by examining the evolution of a single gene. That’s the domain of classical population genetics.
For the next few weeks we’re going to be exploring a field that’s older than classical population genetics, although the approach we’ll be taking to it involves the use of population genetic machinery.2 If you know a little about the history of evolutionary biology, you may know that after the rediscovery of Mendel’s work in 1900 there was a heated debate between the “biometricians” (e.g., Galton and Pearson) and the “Mendelians” (e.g., de Vries, Correns, Bateson, and Morgan).
Biometricians asserted that the really important variation in evolution didn’t follow Mendelian rules. Height, weight, skin color, and similar traits seemed to
vary continuously,
show blending inheritance, and
show variable responses to the environment.
Since variation in such quantitative traits seemed to be more obviously related to organismal adaptation than the “trivial” traits that Mendelians studied, it seemed obvious to the biometricians that Mendelian geneticists were studying a phenomenon that wasn’t particularly interesting.
Mendelians dismissed the biometricians, at least in part, because they seemed not to recognize the distinction between genotype and phenotype. It seemed to at least some Mendelians that traits whose expression was influenced by the environment were, by definition, not inherited. Moreover, the evidence that Mendelian principles accounted for the inheritance of many discrete traits was incontrovertible.
Woltereck’s experiments on Daphnia helped to show that traits whose expression is environmentally influenced may also be inherited. He introduced the idea of a norm of reaction to describe the observation that the same genotype may produce different phenotypes in different environments (Figure 1). When you fertilize a plant, for example, it will grow larger and more robust than when you don’t. The phenotype an organism expresses is, therefore, a product of both its genotype and its environment.
https://en.wikipedia.org/w/index.php?curid=3925138,
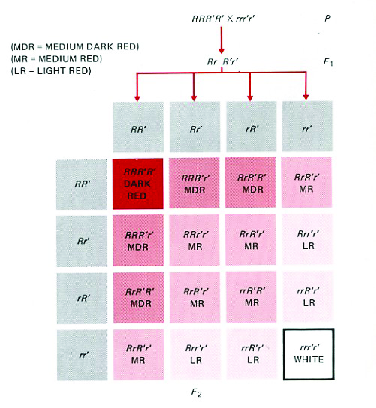
accessed 9 April 2017).Nilsson-Ehle’s experiments on inheritance of kernel color in wheat showed how continuous variation and Mendelian inheritance could be reconciled (Figure 2). He demonstrated that what appeared to be continuous variation in color from red to white with blending inheritance could be understood as the result of three separate genes influencing kernel color that were inherited separately from one another. It was the first example of what’s come to be known as polygenic inheritance. Fisher , in a paper that grew out of his undergraduate Honors thesis at Cambridge University, set forth the mathematical theory that describes how it all works. That’s the theory of quantitative genetics, and it’s what we’re going to spend the rest of the course discussing.
http://www.biology-pages.info/Q/QTL.html,
accessed 9 April 2017).Woltereck’s ideas force us to realize that when we see a phenotypic difference between two individuals in a population there are three possible sources for that difference:
The individuals have different genotypes.
The individuals developed in different environments.
The individuals have different genotypes and they developed in different environments.
This leads us naturally to think that phenotypic variation consists
of two separable components, namely genotypic and environmental
components.3 Putting that into an equation
There’s a surprisingly subtle and important insight buried in that very simple equation: Because the expression of a quantitative trait is a result both of genes involved in that trait’s expression and the environment in which it is expressed, it doesn’t make sense to say of a particular individual’s phenotype that genes are more important than environment in determining it. You wouldn’t have a phenotype without both. At most what we can say is that when we look at a particular population of organisms in a particular environment, some fraction of the phenotypic variation they exhibit is due to differences in the genes they carry and that some fraction is due to differences in the environment they have experienced.6 If we have two individuals with different phenotypes, e.g., Ralph is tall and Harry is short, we can’t even say whether the difference between Ralph and Harry is because of differences in their genes or differences in their developmental environment.
One important implication of this insight is that much of the “nature vs. nurture” debate concerning human intelligence or human personality characteristics is misguided. The intelligence and personality that you have is a product of both the genes you happened to inherit and the environment that you happened to experience. Any differences between you and the person next to you probably reflect both differences in genes and differences in environment. Moreover, even if the differences between you and your neighbor are due to differences in genes, it doesn’t mean that those differences are fixed and indelible. You may be able to do something to change them.
Take phenylketonuria, for example. It’s a condition in which individuals are homozygous for a deficiency that prevents them from metabolizing phenylalanine (https://medlineplus.gov/phenylketonuria.html). If individuals with phenylketonuria eat a normal diet, severe intellectual disabilities can result by the time an infant is one year old. But if they eat a diet that is very low in phenylalanine, their development is completely normal. In other words, clear genetic differences at this locus can lead to dramatic differences in cognitive ability, but they don’t have to.
It’s often useful to talk about how much of the phenotypic variance
is a result of additive genetic variance or of genetic variance.
As you’ll see in the coming weeks, there’s a lot of stuff hidden behind these simple equations, including a lot of assumptions. But quantitative genetics is very useful. Its principles have been widely applied in plant and animal breeding for more than a century, and they have been increasingly applied in evolutionary investigations in the last forty years.10.
Before we worry about how to estimate any of those variance components I just mentioned, we first have to understand what they are. So let’s start with some definitions (Table 1).11
| Genotype | |||
|---|---|---|---|
| Frequency | |||
| Genotypic value | |||
| Additive genotypic value |
You should notice something rather strange about Table 1 when you look at it. I motivated the entire discussion of quantitative genetics by talking about the need to deal with variation at many loci, and what I’ve presented involves only two alleles at a single locus. I do this for two reasons:
It’s not too difficult to do the algebra with multiple alleles at one locus instead of only two, but it gets messy, doesn’t add any insight, and I’d rather avoid the mess.
Doing the algebra with multiple loci involves a lot of assumptions, which I’ll mention when we get to applications, and the algebra is even worse than with multiple alleles.
Fortunately, the basic principles extend with little modification to multiple loci, so we can see all of the underlying logic by focusing on one locus with two alleles where we have a chance of understanding what the different variance components mean.
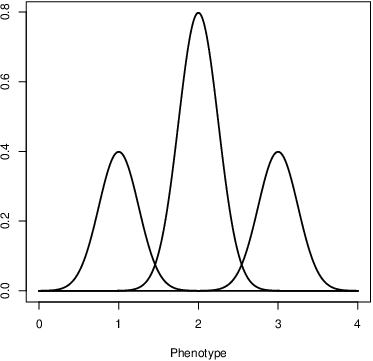
Two terms in Table 1 will almost certainly be unfamiliar to you: genotypic value and additive genotypic value. Of the two, genotypic value is the easiest to understand (Figure 3). It simply refers to the average phenotype associated with a given genotype.12 The additive genotypic value refers to the average phenotype associated with a given genotype, as would be inferred from the additive effect of the alleles of which it is composed. That didn’t help much, did it? That’s because I now need to tell you what we mean by the additive effect of an allele.13
In constructing Table 1 I
used the quantities
The objective is to find values for
Now the first term in square brackets is just the mean phenotype in
the population,
Now divide the first equation in ([eq:zeros]) by
Let’s assume for the moment that we can actually measure the
genotypic values. Later, we’ll relax that assumption and see how to use
the resemblance among relatives to estimate the genetic components of
variance. But it’s easiest to see where they come from if we assume that
the genotypic value of each genotype is known. If it is then, writing
There’s another way to write the expression for
We’ve been through a lot of algebra by now. Let’s run through a couple of numerical examples to see how it all works. For the first one, we’ll use the set of genotypic values in Table 2.
| Genotype | |||
| Genotypic value | 100 | 50 | 0 |
For
For the second example we’ll use the set of genotypic values in Table 3.
| Genotype | |||
| Genotypic value | 100 | 80 | 0 |
For
To test your understanding, it would probably be useful to calculate
These notes are licensed under the Creative Commons Attribution License. To view a copy of this license, visit or send a letter to Creative Commons, 559 Nathan Abbott Way, Stanford, California 94305, USA.